2020
Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale.
Nat Genet. 2020;52(9):969-983.
Rare-variant association testing
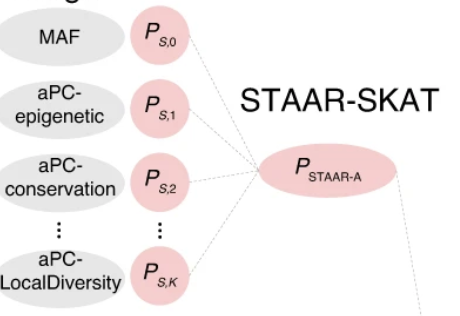
STAAR and its extensions use functional annotation to improve rare-variant association analysis for whole-genome sequencing studies.
Overview
The STAAR family of methods combines statistical genetics and functional genomics so rare variants can be tested in biologically meaningful sets. The ecosystem spans single-study analysis, biobank-scale pipelines, meta-analysis, multi-trait analysis, single-cell-informed noncoding tests, and time-to-event outcomes.
Publications
Representative publications connected to this project.
Nat Genet. 2020;52(9):969-983.
Nat Methods. 2022;19:1599-1611.